Documentations
Protein turnover, Proteomics, Mass spectrometry
Basics
RIANA has three components — integrate, fit, and rollup — called as riana integrate, riana fit, and riana rollup, plus a desktop GUI launched with riana gui. For the help message of each component, type riana <command> --help.
Integrate
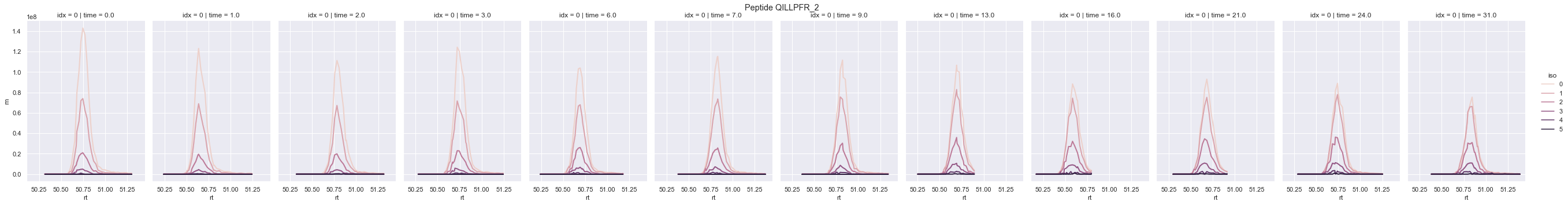
The integrate component reads mass spectrometry data files (.mzML) together with peptide identifications. Identifications come from quantms mzTab (DDA) or DIA-NN report.parquet (DIA) keyed off an SDRF sample sheet (--sdrf), or — for a single mzML — directly from a Percolator target.psms.txt.
RIANA filters identifications on the -q (q-value) threshold. For each qualifying peptide-charge concatamer (including each distinct modified peptidoform), RIANA calculates the accurate mass and m/z of the unlabeled (m0) isotopomer and of each successive isotopomer specified by -i (default 0 1 2 3 4 5, the m0–m5 envelope the D2O fit consumes).
RIANA then locates the peptide in the mzML and extracts the intensity-over-retention-time of each isotopomer. By default it centres a narrow window on the chromatographic apex (--peak-rt apex, --integration-half-width 0.15) and integrates the area by the trapezoid method — replacing the older fixed-width retention-time rectangle, which is still available via --peak-rt ms2 --integration-half-width 1.0. On the SDRF path one identity-stamped <run>_riana.txt is written per run, along with a stage-aware riana_manifest.tsv that chains the downstream steps.
The intensity-over-rt data prior to integration can also be saved with the -w flag.
Fit
The fit component takes in the integration results from multiple mass spectrometry experiments, and fits the relative isotope abundance data across labeling time points to a specified kinetic model.
Three models ar ecurrently supported. In the simple model, the fraction of new protein over labeling time behavior of a peptide is described by an exponential decay equation with one parameter (\(k\)):
\[ A_t = A_{t=0} + (A_{t\rightarrow \infty} - A_{t=0}) \cdot 1 - e^{-k t} \]
where \(A_{t=0}\) is the initial relative abundance of the m0 peak over the mi peaks, and is set to 1 in amino acid labeling experiments. \(A_{t\rightarrow \infty}\) is calculated based on the plateau relative isotope abundance, which is based on the number of labeling sites in a peptide and the precursor plateau enrichment. The latter is supplied through the -r argument in riana fit.
The two-compartment model is implemented as in Guan et al. 2012. The fraction of new protein over labeling time behavior of a peptide is described by a two-exponent model with the protein turnover rate constant (\(k\)) as well as the precursor availability rate constant (\(k_p\)):
\[ A_t = A_{t=0} + (A_{t\rightarrow \infty} - A_{t=0}) \cdot \frac{1 - ( e^{-kt}k_p - e^{-k_pt}k)}{k_p - k } \]
The Fornasiero model two-compartment, three-exponent model additionally attempts to account for the effect of precursor reutilization from proteome-wide protein degradation. The two additional parameters --kr and --rp must be provided to denote the reutilization rate constant and the proportion of protein-bound vs. free precursors. Details are described in the original publication.
Fractional synthesis is solved against the full m0–m5 isotopomer envelope using an IsoSpec forward model: RIANA estimates each peptide’s effective number of labeling sites (Spep) from a per-amino-acid coefficient table and fits θ as the mixture of the natural-abundance and fully-labeled envelopes that best matches the observed isotopomer pattern. The per-amino-acid table is supplied with --coefficients — a bundled preset (commerford literature values, or the ac16 / ipsc / cm cell-line calibration tables) or a path to your own (amino_acid, coefficient) CSV. This replaces the earlier fixed-labeling-site analytic calculation, which biased recovered rate constants low.
Rollup
The rollup component aggregates peptide-level fits to protein-level turnover, reading from the project manifest. Shared peptides are resolved by parsimony (--parsimony unique|isoform), and protein rate constants are estimated by an inverse-variance-weighted per-timepoint collapse (--method weighted) or a pooled fit (--method pooled). With --model "linear simple", RIANA instead fits turnover in φ = log(1 − θ) space and, for a two-condition design, reports a per-protein Δk with a shared-variance p-value and Benjamini–Hochberg adjustment — the cross-condition turnover test.
Variable-modification peptidoforms are integrated at their own m/z and isotopomer envelope. Phospho and side-chain (K-)acetyl peptidoforms roll up as distinct proteoforms (e.g. P12345_pS235, P12345_acK106); constitutive or chemical modifications (protein N-terminal acetyl, methionine oxidation) fold into the bare protein, with oxidized and unoxidized forms merged onto one turnover curve since oxidation does not reset the labeling clock.
All Options
The option lists below are summarized; run riana <command> --help for the authoritative, complete set (the integrate help groups power-user dials under an Advanced integration panel and the match-between-runs options under a Match-between-runs (MBR) panel).
RIANA integrate
riana integrate MZML_PATH ID_PATH [OPTIONS]
MZML_PATH folder containing the mzML file(s)
ID_PATH mzTab (DDA) / DIA-NN report.parquet (DIA) with --sdrf, else a Percolator psms.txt
--sdrf PATH SDRF sample sheet — the primary, identity-keyed intake path
-s, --sample TEXT sample name for the no-SDRF Percolator path (must end in a number) [time0]
-i, --iso TEXT isotopomers to integrate, comma/space separated [0 1 2 3 4 5]
-q, --q_value FDR integrate only PSMs below this q-value [0.01]
-m, --mass_tol PPM ±ppm mass tolerance; taken from the SDRF when present [10]
--peak-rt TEXT window anchor: apex (default), ms2 (0.9.0 parity), or consensus
--integration-half-width MIN|auto integration half-width in RT min [0.15], or 'auto'
-W, --workers N runs integrated concurrently on the --sdrf path [1]
--mbr enable gated match-between-runs (DDA path)
-o, --out PATH output directory [.]
# Advanced: --baseline, --apex-selection, --apex-search-half-width, --extraction_half_width,
# --smoothing (+ --smoothing-polyorder), --mass_difference, --ppm-alert, --prominence-k,
# --width-rel-height, --apex-n-consensus, --no-rt-check (+ --scan-rt-tol), -w/--write_intensities
# To reproduce 0.9.0: --peak-rt ms2 --integration-half-width 1.0RIANA fit
riana fit [RIANA_PATH ...] [OPTIONS]
RIANA_PATH one or more <run>_riana.txt files (single-mzML path); or use --manifest
--manifest PATH the project manifest from integrate --sdrf (groups runs into curves)
--coefficients TEXT REQUIRED per-amino-acid Spep table: preset (commerford|ac16|ipsc|cm) or a CSV path
-m, --model TEXT simple | guan | fornasiero [simple]
-l, --label TEXT hw (heavy water, default) | o18 (in progress)
--kp / --kr / --rp fixed precursor parameters for the two-compartment models
-q, --q_value FDR fit only points below this q-value [0.01]
-d, --depth INT fit only peptidoforms seen at >= this many distinct labeling timepoints [3]
-r, --ria FLOAT precursor enrichment (RIA max), e.g. 0.06 for 6% D2O [0.06]
--exclude-mbr drop match-between-runs points before fitting
-W, --workers N process-level parallelism [1]
-o, --out PATH output directory [.]RIANA rollup
riana rollup [FIT_DIR] [OPTIONS]
FIT_DIR directory of fit outputs; or use --manifest
--manifest PATH the project manifest (roots outputs at the project dir)
--parsimony TEXT unique (default) | isoform — shared-peptide attribution
--method TEXT weighted (inverse-variance per-timepoint collapse) | pooled
-m, --model TEXT simple | guan | fornasiero | "linear simple"
--reference-condition TEXT reference condition for the linear-simple Δk contrast
--phi-limit FLOAT plateau-truncation threshold in φ-space (linear simple only) [-4]
--min-peptides / --min-points / --min-r2 admission gates
-W, --workers N process-level parallelism [1]
-o, --out PATH output directory [.]